




Papulosis linfomatoide

Néstor P. Sánchez, MD, FAAD, FADA
Profesor de Dermatología y Patología

Carlos Sarriera Lazaro, MD
Introducción
La papulosis linfomatoide es una erupción cutánea caracterizada por pápulas y nódulos recurrentes que sanan por sí mismos. Tiene un curso crónico que puede durar meses o años.
A nivel histológico, hay células T atípicas con proliferación linfoide CD30+ que podrían confundirse con un linfoma maligno. El pronóstico de la condición es excelente, aunque se ha visto que estos pacientes tienen un riesgo aumentado de desarrollar un linfoma maligno.
Epidemiologia y patogénesis
La papulosis linfomatoide es una condición rara (en los Estados Unidos se estima entre 1,2 y 1,9 casos por millón de personas), puede ocurrir a cualquier edad, con mayor incidencia después de los 50 años. Entre un 5 y un 50% se puede relacionar con un linfoma maligno. Su etiología aún se desconoce, pero se asocia a una sobreexpresión de CD30 en las células T atípicas que resulta en su proliferación, ocasionando las lesiones características. Una hipótesis es que la eventual regresión de estas lesiones se debe a un mecanismo de apoptosis aumentado en estas células atípicas.
Presentación clínica
Las lesiones clásicas de la papulosis linfomatoide son pápulas y nódulos eritematosos o violáceos, que luego desarrollan una hemorragia central, necrosis y costra (Figura 1). Estas coexisten en diferentes estadios de desarrollo y son clínicamente indistinguibles de las lesiones del linfoma anaplásico de células grandes.
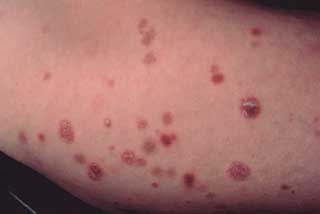
Después, desaparecen espontáneamente en un periodo de 2 a 8 semanas. Su curso es crónico y recurrente y puede durar hasta más de 40 años. Las lesiones no ocurren en ningún lugar en particular, pero usualmente se encuentran en las extremidades, manos, cara y genitales. Raramente aparecen en las membranas mucosas.
Diagnóstico
Clínicamente, se debe sospechar la papulosis linfomatoide en aquellos pacientes que presentan una erupción de lesiones papulares y nodulares recurrentes, en diferentes estadios de desarrollo y que sanan espontáneamente en ausencia de síntomas constitucionales. Para confirmar el diagnóstico, es necesaria una biopsia de piel. Se recomienda que se haga una excisión completa de 2 o más de las lesiones inflamatorias que no demuestren evidencia de necrosis o involución.
Histológicamente, la papulosis linfomatoide varía de acuerdo con el estadio de la lesión. Se ha reportado que existen 6 tipos histológicos mayores de esta condición, siendo el tipo A el más común (representa el 75% de los casos). Se caracteriza por un infiltrado denso, superficial y profundo dermal (perivascular e intersticial) en forma de cuña de células T atípicas entremezcladas con linfocitos pequeños, neutrófilos, eosinófilos e histiocitos. Se pueden apreciar varias mitosis que podrían ser atípicas. El patrón histológico tipo A es muy similar al infiltrado polimórfico del linfoma de Hodgkin. Un análisis inmunohistoquímico revela la expresión de CD30 y otros antígenos relacionados al linfoma de Hodgkin en estas células T atípicas grandes. La evaluación molecular demuestra un reordenamiento clonal de genes TCR en 40 a 100% de los casos de papulosis linfomatoide.
Hacer el diagnóstico de papulosis linfomatoide puede ser difícil, ya que la histología es altamente variable dependiendo al estadio de la lesión y puede solapar –al igual que la presentación clínica– la apariencia del linfoma anaplásico de células grandes. Es por esto por lo que es indispensable llevar a cabo una correlación clinicopatológica cuando se sospecha esta condición, para así llegar a un diagnóstico certero y establecer un plan de tratamiento adecuado.
Diagnóstico diferencial
El diagnóstico diferencial de papulosis linfomatoide incluye: linfoma cutáneo primario anaplásico de células grandes, micosis fungoide, pitiriasis liquenoide y varioliforme aguda (PLEVA), pitiriasis liquenoide crónica (PLC), foliculitis, granuloma de Majocchi, picada de artrópodo y sarna.
Tratamiento
Existen varias modalidades de tratamiento para la papulosis linfomatoide que pueden acelerar la resolución de las lesiones existentes y prevenir el desarrollo de nuevas. Sin embargo, ninguna terapia altera el curso natural de la condición ni previene el desarrollo de un linfoma asociado.
Para pacientes con enfermedad limitada, se puede usar esteroides tópicos súper-potentes. Para pacientes con enfermedad extensa y sintomática, se sugiere usar metotrexato oral o subcutáneo en dosis baja (5 a 35 mg por semana) con ácido fólico oral (1 mg diario) como terapia inicial. El metotrexato es un tratamiento efectivo para suprimir la condición. Sin embargo, al dejar de tomarlo, la misma recurre en un periodo de 1 a 2 semanas.
En aquellos pacientes que no respondan a metotrexato o con una contraindicación al mismo, se recomienda terapia con psoraleno oral con radiación ultravioleta A (PUVA) 2 veces por semana por 6 a 8 semanas o hasta que se vayan las lesiones. Para pacientes pediátricos con enfermedad extensa, se recomienda el uso de esteroides tópicos o fototerapia con radiación ultravioleta B de banda estrecha (UVB). Existe evidencia limitada de otros posibles tratamientos para esta condición, tales como terapia fotodinámica, retinoides orales o tópicos, carmustina tópica, entre otros, que han demostrado tener algún efecto de supresión de la condición en estudios pequeños o en reportes de caso.
Linfomas asociados
En estudios recientes, se ha visto que más del 50% de los pacientes con papulosis linfomatoide desarrollan un linfoma maligno antes, durante o después de tener la condición. Los linfomas más comunes son micosis fungoide, linfoma anaplásico cutáneo o nodal de células grandes y linfoma de Hodgkin. No se han establecido criterios o hallazgos particulares que puedan predecir la progresión de papulosis linfomatoide a un linfoma maligno. Por esto, es muy importante que el médico evalúe a su paciente por lo menos 2 veces al año y que lo instruya a evaluarse si comienza a padecer de fiebre, escalofríos, sudoración nocturna, malestar general o pérdida de peso, si le salen lesiones papulares o nodulares que no se resuelven por sí solas en menos de 2 meses o si se percatan de nódulos linfáticos agrandados en el cuello, área inguinal o en las axilas.
Comentario
La papulosis linfomatoide se puede confundir clínicamente e histológicamente con otras entidades benignas y malignas de la piel. Es por esto que es esencial hacer una correlación clínica completa cuando se sospeche esta condición para confirmar el diagnóstico, tratar al paciente con prontitud y vigilar que la enfermedad no progrese a una malignidad.
Referencias
- Bekkenk MW et al. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders. Blood. 2000;95(12):3653-61.
- Bolognia JL, Schaffer JV, Duncan KO, Ko CJ. Dermatology Essentials. Elsevier Saunders, 2014.
- Braun FK, Hirsch B, Al-Yacoub N, Durkop H, Assaf C, Kadin ME, et al. Resistance of cutaneous anaplastic large-cell lymphoma cells to apoptosis by death ligands is enhanced by CD30-mediated overexpression of c-FLIP. J Invest Dermatol. 2010;130(3):826-40.
- Kadin M, Nasu K, Sako D, Said J, Vonderheid E. Lymphomatoid papulosis. Am J Pathol. 1985;119(2):315-25.
- Kadin ME. Lymphomatoid papulosis. UpToDate. 23 May 2016.
- Kempf W. CD30+ lymphoproliferative disorders. J Cutan Pathol. 2006;33 Suppl 1:58-70.
- Kempf W et al. Lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118(15):4024-35.
- Kunishige JH, McDonald H, Alvarez G, Johnson M, Prieto V, Duvic M. Lymphomatoid papulosis and associated lymphomas: A retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34(5):576-81.
- Macaulay WL. Lymphomatoid papulosis. Arch Derm. 1968;97(1):23-30.
- Sanchez NP, Pittlkow MR, Muller SA, Banks PM, and Winkelmann RK. The clinicopathologic spectrum of lymphomatoid papulosis: Study of 31 cases. J Am Acad Dermatol. 1983;8(1):81-94.
- Steinhoff M, Hummel M, Anagnostopoulos I, Kaudewitz P, et al. Single-cell analysis of CD30+ cells in lymphomatoid papulosis demonstrates a common clonal T-cell origin. Blood. 2002;100(2):578-84.
- Wang HH, Myers T, Lach LJ, Hsieh CC, Kadin ME. Increased risk of lymphoid and nonlymphoid malignancies in patients with lymphomatoid papulosis. Cancer. 1999;86(7):1240-5.
- Wieser I, Oh CW, Talpur R, Duvic M. Lymphomatoid papulosis. J Am Acad Dermatol. 2016;74(1):59-67.
- Willemze R et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105(10):3768-85.
- Zic JA and Raugi GJ. Lymphomatoid papulosis. Medscape. Last updated on 04 Oct 2017.



