Vasculitis urticarial:
Correlación clínico-patológica

Néstor Sánchez, MD, FAAD
Profesor de Dermatología y Patología
Escuela de Medicina, Universidad de Puerto Rico
Consultor, Hospital Menonita, Aibonito

Luis Enrique IV Santaliz-Ruiz, MD
Aspectos clínicos
La vasculitis urticarial (VU) es una entidad clínico-patológica rara. Se presenta con lesiones cutáneas caracterizadas por ronchas eritematosas, pruríticas y elevadas, similares a la urticaria. En pacientes con episodios recurrentes, se puede confundir con urticaria crónica, pero las ronchas de la VU persisten más de 24 horas y muestran una histopatología de vasculitis leucocitoclástica.
Las lesiones pueden arder o doler y ocurren diaria o mensualmente. La mayoría se desvanecen sin dejar rastro, aunque a veces dejan una hiperpigmentación residual. Su severidad va desde lesiones leves hasta compromiso sistémico (angioedema de labios, ojos, faringe y hasta púrpuras palpables y petequias en las extremidades inferiores, poliartralgia o poliartritis, dolor abdominal/de pecho, fiebre, hemoptisis, uveitis, episcleritis, microhematuria y microproteinuria).
Epidemiología
Cerca del 3% al 5% de los pacientes evaluados por una supuesta urticaria crónica tienen realmente VU. Suele presentarse en mujeres jóvenes y de mediana edad, y es menos común en hombres. Es raraen niños (solo hay 5 casos pediátricos en la literatura médica). Su mayor incidencia es en la cuarta década de vida. Los síntomas tienen una duración promedio de 3 años, pudiendo llegar a 23 años.
Patofisiología
El mecanismo de la VU comienza con el desarrollo de anticuerpos circulantes (IgG o IgM) contra autoantígenos (C1 precipitina), o contra antígenos exógenos como medicamentos o agentes infecciosos (hepatitis B o C). Los anticuerpos se adhieren a los antígenos y forman inmunocomplejos circulantes que se depositan en la pared de los vasos sanguíneos de pequeño calibre. Esto provoca la activación de complemento por la vía clásica y lleva a la generación de anafilatoxinas. Estas inducen la desgranulación de mastocitos y a la síntesis de novo de citoquinas y quimioquinas –responsables del aumento en la permeabilidad capilar–, la formación de ronchas, el depósito de inmunocomplejos y la quimiotaxis de neutrófilos que fagocitan escombros en la membrana basal de los vasos sanguíneos y liberan enzimas proteolíticas que ocasionan más edema y daño tisular.
Pruebas de laboratorio
Se debe realizar una batería de pruebas de laboratorio en pacientes con sospecha de VU: conteo completo de células sanguíneas (CBC), pruebas de función renal, análisis de enzimas hepáticas, niveles de CH50, C3, C4, C1q y anti-C1q, urianálisis, tasa de sedimentación eritrocítica (ESR), pruebas de virus de hepatitis B y hepatitis C, factor reumatoideo (RF), anticuerpos anti-ADN (anti-dsDNA) y anticuerpos anti-péptidos cíclicos citrulinados. Los pacientes con VU suelen tener elevación en ESR, niveles bajos de complemento en suero y ANA positivos. Los niveles de ESR no se correlacionan con la severidad de VU, ni con el compromiso sistémico. En la VU sistémica, la hipocomplementemia tiene significado pronóstico.
Clasificación
La VU se clasifica en 3 grupos (según manifestación sistémica y nivel de complemento): normo-, e hipocompleméntica y síndrome hipocompleméntico (VUN, VUH y SVUH). En la VUN, los niveles de complemento son normales y el pronóstico es mejor ya que el compromiso sistémico es mínimo o nulo (más allá de ronchas, angioedema, fiebre y púrpuras). En la VUH, los niveles de complemento son bajos (C1q, C3, C4, CH50) y hay ronchas y angioedema, fiebre, artralgias/artritis, dolor abdominal, náusea o diarreas y cierta implicación sistémica. Su pronóstico es menos alentador que en VUN. El SVUH es más severo que la VUH, pues los pacientes presentan mayor compromiso sistémico (más allá de ronchas y angioedema). Además, desarrollan glomerulonefritis con hematuria y proteinuria, EPOC aúun sin fumar y a una edad muy temprana (30-40 años), pericarditis, valvulopatías, pseudotumor cerebri, mononeuritis, episcleritis o uveítis. A pesar de su efecto multiorgánico, rara vez los pacientes perecen como consecuencia intrínseca a la VU.
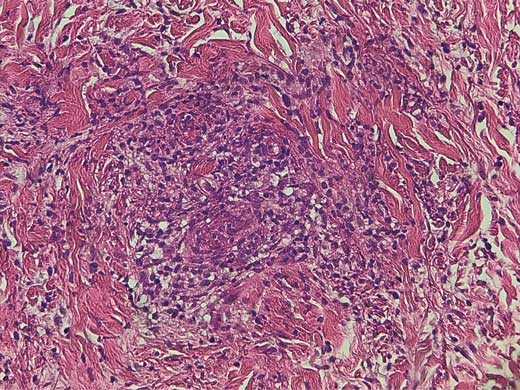
Diagnóstico histopatológico
Ante la dificultad para distinguir clínicamente la VU de la urticaria alérgica recurrente, una biopsia de las lesiones activas es el estándar de oro para el diagnóstico definitivo. La histopatología de la VU se caracteriza por una vasculitis leucocitoclástica con inflamación perivascular y en las paredes de capilares y vénulas. Los neutrófilos con fragmentación nuclear (leucocitoclasia) predominan en el infiltrado inflamatorio, aunque a veces también puede haber eosinófilos. Además, hay edema de las células endoteliales, degeneración fibrinoide de la pared vascular y extravasación perivascular e intersticial de eritrocitos. El edema e infiltrado inflamatorio ocurren también en el colágeno dermal.
Las pruebas de inmunofluorescencia demuestran depósitos granulares de IgG, IgM, C3 y/o fibrinógeno en las paredes de vasos sanguíneos y la membrana basal epidermal. Por otro lado, la urticaria crónica alérgica muestra un infiltrado perivascular linfo-histiocítico (vasculitis linfocítica). Las células endoteliales pueden edematizarse (raramente extravasan, y si las células inflamatorias infiltran la dermis puede haber una leve espongiosis epidermal). Puede haber infiltrados de neutrófilos y eosinófilos, pero la leucocitoclasia y la degeneración fibrinoide están ausentes. En urticaria crónica es negativa la tinción para inmunoglobulinas y complementos. La urticaria aguda se distingue al microscopio de la VU ya que la dermis muestra un edema moderado a severo, con escaso infiltrado inflamatorio y sin vasculitis.
Enfermedades asociadas a VU
Las lesiones de VU ocurren a veces en el contexto de enfermedades como lupus sistémico eritematoso (LSE), síndrome de Sjögren, hepatitis B o C, virus de cocksakie B, mononucleosis infecciosa, enfermedad de Lyme, mieloma IgA, crioglobulinemia y enfermedad del suero. Estos pueden desencadenar formación y depósitos de inmunocomplejos, lo que es consistente con la patogénesis idiopática de la VU. De hecho, se especula que la VU es parte del espectro de LSE pues se han visto casos de VUH que progresan a lupus sistémico y viceversa. VUH y LSE son mediados por inmunocomplejos y tienen características comunes: artritis/artralgias, uveítis, episcleritis, pleuritis, glomerulonefritis, anticuerpos antinucleares (ANA), anticuerpos anti-ADN de doble cadena (anti-dsDNA), Sin embargo, su relación patogénica permanece incógnita.
Opciones terapéuticas
El tratamiento contra la VU depende de su severidad. Los antihistamínicos sirven para el alivio sintomático del prurito en casos leves sin compromiso sistémico, pero no alteran el curso de la enfermedad ni impactan la inflamación ocasionada por los inmunocomplejos. La indometacina oral es una alternativa en casos leves. Por lo general, la VU es resistente a agentes antiinflamatorios no esteroidales y a antihistamínicos. Algunos pacientes con enfermedad leve limitada a la piel requieren breves terapias con corticosteroides orales como prednisona (1mg/kg/d). La misma dosis se prescribe en exacerbaciones periódicas en enfermedad leve, moderada y severa. Una vez surge una mejoría significativa, comienza el destete seriado de la prednisona. La VU moderada con cierta enfermedad sistémica también requiere prednisona (igual dosis) y una transición a otros agentes inmunomoduladores (dapsona, colchicina y la hidroxicloroquina) con menos efectos adversos que los corticosteroides a largo plazo. La VU severa necesita de otros fármacos inmunosupresores como micofenolato de mofetilo, metotrexato, azatioprina o ciclosporina. Los casos recalcitrantes que no responden adecuadamente a estos medicamentos pudieran beneficiarse de agentes biológicos con resultados prometedores en fases experimentales (omalizumab, rituximab, anakinra o canakinumab).
Referencias -# Sanchez NP, Winkelmann RK, Schroeter AL, Dicken CH. The clinical and histopathologic spectrums of urticarial vasculitis: study of forty cases. J Am Acad Dermatol. 1982;7(5):599-605. -# Venzor J, Lee WL, Huston DP. Urticarial vasculitis. Clin Rev Allergy Immunol. 2002;23(2):201-216. -# Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28(1):97-100. -# Ghazanfar MN, Thomsen SF. Omalizumab for Urticarial Vasculitis: Case Rep Dermatol Med. 2015:576893. -# Fueyo-Casado A, Campos-Muñoz L, González-Guerra E, Pedraz-Muñoz J, Cortés-Toro JA, López-Bran E. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. 2017; 42:403-405. -# Hamad A, Jithpratuck W, Krishnaswamy G. Urticarial vasculitis and associated disorders. Ann Allergy Asthma Immun. 2017;118(4):394-8.