Amiloidosis cardiaca:
Causa de insuficiencia cardiaca

Karen Rodríguez Maldonado, MD, FACC
Especialista en Cardiología
Presidente del Comité Científico del
Colegio Americano de Cardiología-Capítulo de Puerto Rico
Miembro de la Sociedad Puertorriqueña de Cardiología
Los pacientes que presentan signos y síntomas de insuficiencia cardiaca frecuentemente son evaluados para detectar causas como la enfermedad obstructiva de las arterias coronarias, la enfermedad valvular, las arritmias y las cardiomiopatías dilatadas secundarias a otras causas. Sin embargo, no es así para descartar las condiciones infiltrativas que afectan al corazón. Estas enfermedades infiltrativas pueden comprometer la elasticidad normal del miocardio provocando insuficiencia cardiaca (IC). La amiloidosis cardiaca (CA) es una de estas condiciones que a menudo puede pasar desapercibida.
Definición de amiloidosis cardiaca y manifestación clínica
La amiloidosis es el término general que utilizamos para describir cuando un precursor de proteína con estructura inestable se desdobla y se agrega en formas insolubles conocidas como las fibrillas de amiloide. Estas fibrillas se pueden depositar en el espacio extracelular de órganos sólidos o de tejidos blandos. A la histología, los tejidos afectados se caracterizan por una birrefringencia verde manzana con luz polarizada al ser teñidos con el tinte rojo congo.
Se conocen al menos 38 diferentes precursores de fibras de amiloide humano. Estas pueden afectar cualquier órgano o tejido del cuerpo. La formación de estas fibrillas insolubles puede ser causada por mutaciones genéticas, infecciones, malignidades, enfermedades autoinmunes y la hemodiálisis, entre otras.
La manifestación clínica y cómo se clasifica la condición dependen del tipo de precursor de proteína y del tejido afectado. En la amiloidosis cardiaca (CA), el amiloide se deposita en el tejido extracelular del corazón y la gran mayoría de los casos (más del 95%) son de dos tipos: amiloidosis por transtiretina y amiloidosis por cadena liviana.
Amiloidosis cardiaca por transtiretina (ATTR)
La transtiretina (TTR) es una proteína sintetizada por el hígado que transporta la hormona tiroidea y el retinol. En la amiloidosis cardiaca, la TTR se disocia formando las fibrillas de amiloide. Existen dos formas de amiloidosis por TTR: la salvaje (w-ATTR) y la heredada (h-ATTR). La ATTR se presenta mayormente en pacientes mayores de 70 años.
La literatura científica sugiere que la forma salvaje es una causa de IC no diagnosticada en el 13% de los casos de fallo cardiaco con función sistólica preservada, y en el 16% de los casos con estenosis aórtica que requieren reemplazo por catéter.
En cuanto a la forma heredada, se han podido identificar más de 120 mutaciones. Algunas variantes genéticas son endémicas en ciertas regiones de Portugal, Suiza, Japón, Brasil, Italia y España. Otras se han podido identificar en descendientes de africanos y afrocaribeños.
Los pacientes con ATTR comúnmente desarrollan el síndrome de túnel carpal bilateral, que se puede presentar años antes de la aparición de los síntomas cardiacos. Otros síntomas pueden incluir la estenosis del canal lumbar y la ruptura del tendón del bíceps.
Amiloidosis cardiaca por cadenas livianas (AL)
En la AL, las células monoclonales plasmáticas secretan cadenas ligeras que se agregan formando las fibras de amiloide. La amiloidosis cardiaca (CA) por AL tiene peor pronóstico que la ATTR y se puede presentar desde los 40 años. Las manifestaciones clínicas de AL incluyen síntomas no específicos como fatiga, pobre apetito y pérdida de peso. También se pueden presentar enfermedad renal, neuropatía, hepatomegalia, sangrado gastrointestinal, púrpura y macroglosia.
Diagnóstico
Pruebas de laboratorio
Los pacientes con amiloidosis cardiaca (CA) pueden presentar niveles elevados de troponina, de creatinina sérica, de bilirrubina y del péptido natriurético. Cuando se sospecha CA se deben evaluar niveles en sangre de las cadenas livianas kappa y lambda, y hacer electroforesis además de la inmunofijación de proteínas en sangre y orina.
Electrocardiograma
El electrocardiograma (ECG) podría mostrar un QRS con voltaje bajo (más frecuente en AL) aun cuando se esperaría un voltaje alto por la hipertrofía del ventrículo izquierdo. Sin embargo, la ausencia de este hallazgo no excluye el diagnóstico de CA. Los hallazgos más comunes en un análisis de casos con w-ATTR fueron la fibrilación auricular y los patrones de pseudoinfarto, seguidos por un voltaje sugestivo de hipertrofía de ventrículo y por bloqueos en la conducción atrioventricular.
Ecocardiografía
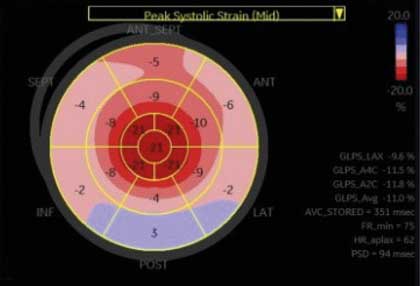
La fracción de eyección del ventrículo izquierdo está preservada en el inicio de la CA, y la disfunción diastólica progresa a medida en que avanza la enfermedad. Los hallazgos más típicos incluyen: engrosamiento de las válvulas y de los ventrículos (generalmente asimétrico en ATTR, exhibiendo el máximo de hipertrofia en el septum: >12 mm), cavidades ventriculares no dilatadas, dilatación auricular, efusión pericárdica y aspecto granular del miocardio. El depósito predominante del amiloide en los segmentos basales y medios del miocardio –y no así en los segmentos apicales– genera una menor deformación miocárdica longitudinal (speckle tracking) en los segmentos más comprometidos.
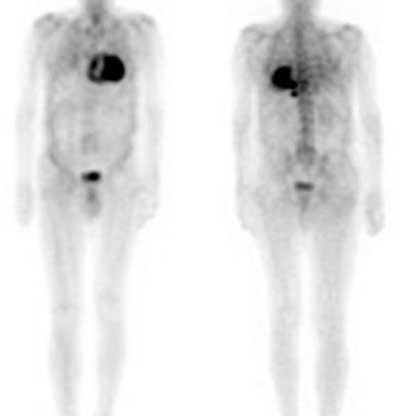
Medicina nuclear
Un estudio de medicina nuclear con pirofosfato (PYP) marcado con Tecnecio 99 puede detectar el depósito de amiloide en el músculo cardiaco. Se compara el nivel de captación PYP en el corazón con el de las costillas, clasificándolo en 3 grados: grado 0 (ausencia de captación cardiaca y captación ósea normal); grado 1 (captación cardiaca inferior a la ósea); grado 2 (captación cardiaca igual a la ósea); y grado 3 (captación cardiaca mayor que la ósea). En el caso de AL, la captación es ausente o grado 1.
Resonancia magnética
Las imágenes de resonancia magnética cardiaca luego del realce tardío con gadolinio pueden ser útiles para diagnosticar CA. Sin embargo, no necesariamente ayudan a distinguir entre AL y ATTR.
Biopsia del músculo cardiaco
La biopsia del músculo cardiaco no se requiere en todos los pacientes si los estudios de imágenes y los análisis de proteína monoclonal nos dirigen en el diagnóstico. La biopsia no se requiere si el paciente tiene una prueba de medicina nuclear grado 2 o 3 en ausencia de proteínas monoclonales, ya que esta información es suficiente para el diagnóstico de ATTR. Tampoco es requerida si el resultado de la resonancia magnética es sugestivo de CA y se confirma con otros análisis el diagnóstico de AL.
Si el resultado de proteína monoclonal es anormal, entonces se requiere la evaluación de un especialista en hematología/oncología. En los casos en que los análisis, las imágenes y las evaluaciones realizadas no sean concluyentes, se podría necesitar una biopsia de grasa y, de esta resultar negativa, requerir una biopsia del músculo del corazón. El tejido obtenido se somete a unas tinciones especiales para confirmar la presencia del amiloide, mientras que el tipo de amiloide se detecta por medio de inmunohistoquímica y el análisis de espectrometría de masa, entre otros.
Tratamiento
El tratamiento de AL consiste en un régimen de quimioterapia y/o de trasplante de células madre autólogas para los pacientes que así califiquen.
Para los pacientes con amiloidosis cardiaca por TTR –ya sea la forma heredada o la forma salvaje– con insuciencia cardiaca clase funcional NYHA I-III, existe un estabilizador de la transtiretina que está asociado a una disminución en la mortalidad cardiovascular y a las hospitalizaciones por insuficiencia cardiaca. Los pacientes con la forma heredada de ATTR deben ser referidos a especialistas de transplante de hígado, ya que esta podría ser una opción para algunos de estos casos.
Conclusión
La amiloidosis cardiaca es una enfermedad progresiva que afecta el funcionamiento normal del corazón provocando una variedad de presentaciones clínicas en los pacientes afectados. Se requiere de varios métodos de análisis y de imágenes del corazón para llegar al diagnóstico adecuado. Una vez que se diagnostica y se trata la condición, es posible ayudar a mejorar la prognosis y la calidad de vida del paciente.
Referencias
- American Society of Nuclear Cardiology (ASNC)/American Heart Association (AHA)/American Society of Echocardiography (ASE)/European Association of Nuclear Medicine (EANM)/Heart Failure Society of America (HFSA)/International Society of Amyloidosis (ISA)/Society for Cardiovascular Magnetic Resonance (SCMR)/Society of Nuclear Medicine and Molecular Imaging (SNMMI): Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis – Part 1 of 2: Evidence base and standardized methods of imaging (2021)
- Brownrigg J, Lorenzini M, Lumley M, Elliott P. Diagnostic performance of imaging investigations in detecting and differentiating cardiac amyloidosis: a systematic review and meta-analysis. ESC Heart Fail 2019; 6:1041.
- Castaño A, Narotsky DL, Hamid N, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017; 38:2879.
- Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol 2016; 68:1323.
- González-López E, Gagliardi C, Dominguez F, et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J 2017; 38:1895.
- González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36:2585.
- Maurer MS et al. Tafamidis Treatment for Patients with Transthyretin Amyloids Cardiomyopathy. NEJM 2018; 379:1007-1016
- Mussinelli R, Salinaro F, Alogna A, et al. Diagnostic and prognostic value of low QRS voltages in cardiac AL amyloidosis. Ann Noninvasive Electrocardiol 2013; 18:271.
- Nativi-Nicolau J, Maurer MS. Amyloidosis cardiomyopathy: update in the diagnosis and treatment of the most common types. Curr Opin Cardiol 2018; 33:571.