









Mieloma múltiple:
Enfermedad antigua, nuevos tratamientos

Justiniano Castro, MD
Hematólogo-Oncólogo
Sección de Hematología-Oncología
Hospital Universitario
Departamento de Medicina
Recinto de Ciencias Médicas de la
Universidad de Puerto Rico
Las malignidades hematológicas constituyen un grupo significativo de todos los tipos de cáncer. El mieloma múltiple (MM) es un tipo de malignidad hematológica en la cual las células plasmáticas sufren una transformación maligna. Las células plasmáticas normales se encuentran en la médula ósea y son una parte importante del sistema inmunológico. Cuando los linfocitos B responden a una infección, maduran y se convierten en células plasmáticas. Las células plasmáticas producen los anticuerpos o inmunoglobulinas que ayudan al cuerpo a atacar y matar los agentes infecciosos. En general, cuando las células plasmáticas se vuelven cancerosas y se producen sin control, se denomina MM. Las células plasmáticas producen una proteína anormal que se conoce por varios nombres diferentes como inmunoglobulina monoclonal, proteína monoclonal (proteína M), pico monoclonal o paraproteína.
El mieloma múltiple constituye el 10% de todas las malignidades hematológicas y el 20% de las muertes producidas por estas. La edad media en que se desarrolla esta enfermedad es de 60 años. Esta condición sigue siendo responsable de un número significativo (2%) de muertes asociadas al cáncer.
Historia
La enfermedad de mieloma múltiple fue descrita por primera vez en 1844 y un año más tarde, en 1845, el Dr. Henry Bence Jones descubrió un material proteínico en la orina que luego se llamó proteína Bence Jones. En 1875 se utilizó por primera vez el nombre de neoplasia de células plasmáticas. El agente quimioterapéutico más activo en MM, el melfalán, se usó por primera vez en 1940. En 1980, se efectuó el primer trasplante de medula ósea autólogo para esta condición. En la última década han surgido múltiples tratamientos innovadores, los que se han desarrollado debido a los avances recientes en el conocimiento de la fisiopatología de la enfermedad.
Fisiopatología
No se conoce exactamente cómo se desarrolla el MM.
Se sabe que hay múltiples factores involucrados, como:
- El microambiente en la medula ósea;
- La angiogénesis; y
- Los cambios genéticos y epigenéticos que son necesarios para que se desarrolle la enfermedad.
Los receptores tipo “Toll” (TLR) son moléculas en la superficie celular que detectan y responden a infecciones microbianas. Recientemente, alteraciones en estos han sido asociadas al desarrollo de mieloma.
El 3% de la población puede tener una elevación clonal de alguna de las inmunoglobulinas después de los 50 años, usualmente de menos de 3 gr/dl. Esto se conoce como gammapatía monoclonal de significado incierto (GMSI). Esta es una condición benigna, asintomática, pero con potencial de transformación a MM a razón del 1% de los pacientes por año. Estos usualmente se evalúan cada 6 meses con pruebas de electroforesis de inmunoglobulinas séricas (EDIS). Se puede concluir que la GMSI es una condición premaligna, pero no se conocen con exactitud los factores específicos que contribuyen a su trasformación maligna.
Existe un pequeño grupo de pacientes que desarrollan una condición intermedia entre GMSI y MM, que se conoce como mieloma indolente (MI). Este se define como una condición con una gammapatia de más de 3gm/dl en suero o en orina, la medula ósea con más de 10% de células plasmáticas, pero sin signos ni síntomas de mieloma (anemia, lesiones óseas etc.). Esta condición ocurre en un pequeño grupo de pacientes y muchas veces no requiere tratamiento.
Podríamos ver la evolución de EDIS a mieloma en el siguiente esquema:

El 75 % de los pacientes con MM presentan anemia, sin encontrarse la etiología en las pruebas de rutina. Algunos de los síntomas comunes son: dolor en huesos (60%), insuficiencia renal (50%), síntomas constitucionales (30%) e hipercalcemia (30%), entre otros (síntomas CRAB).
El diagnóstico de MM se debe sospechar en las siguientes situaciones:
- Dolor en los huesos con lesiones líticas descubiertas en estudios de imagen o radiografías esqueléticas de rutina;
- Aumento de la concentración de proteína sérica total y/o presencia de una proteína monoclonal en la orina o suero (electroforesis/inmunofijación de sangre/orina);
- Signos o síntomas sistémicos sugestivos de malignidad, como anemia donde no se encuentra la causa;
- Hipercalcemia severa, muchas veces sintomática; y
- Insuficiencia renal aguda con un análisis de orina rutinario o, en raras ocasiones, síndrome nefrótico debido a la amiloidosis concurrente de cadena ligera (AL) de inmunoglobulina.
Un grupo significativo de los pacientes con MM no tiene proteínas elevadas en sangre (20%), pero hay un pico monoclonal en orina (cadena liviana libre en orina, –free light chain-). Este grupo de pacientes muchas veces visita al médico por un problema de proteína en orina. Los plasmocitomas extramedulares (EP), que son tumores sólidos llenos de células plasmáticas, pueden ser una presentación de MM (7% de los casos) y se diagnostican mejor mediante tomografía por emisión de positrones (PET) / tomografía computarizada (CT) y/o MRI. La presencia de EP al momento del diagnóstico se asocia con una supervivencia inferior y es uno de los criterios diagnósticos de MM. Otro grupo de pacientes (6%) desarrollará EP más adelante en el curso de la enfermedad, necesitando con frecuencia tratamiento local, como radioterapia.
Para hacer el diagnóstico de MM, necesitamos:
Células plasmáticas de médula ósea ≥10% o plasmocitoma óseo extramedular o probado en biopsia y uno o más de los siguientes eventos que definen el mieloma:
- Evidencia de daño en el órgano terminal que puede atribuirse al trastorno proliferativo subyacente de las células plasmáticas, específicamente: hipercalcemia (calcio sérico > 1 mg/dL más alto que el límite superior de normal o > 11 mg/dL); insuficiencia renal (creatinina > 2 mg/dL); anemia: valor de hemoglobina > 2 g/dL por debajo del límite inferior de lo normal, o un valor de hemoglobina <10 g/dL;
- Lesiones óseas: una o más lesiones osteológicas en la radiografía del esqueleto, CT, MRI o PET-CT;
- Uno o más de los siguientes biomarcadores de malignidad son diagnósticos para MM:
– Células plasmáticas de médula ósea ≥ 60%;
Cadenas livianas (kappa/lambda): relación de cadenas ligeras libres en suero ≥100; y
– Más de una lesión ósea focal en MRI.
Factores pronósticos
En la evaluación de los pacientes de MM, debemos incluir:
Hemograma, pruebas químicas, LDH, beta2-microglobulina, electroforesis de suero y orina, inmunofijación de suero y orina (24 horas), niveles de calcio, albumina, proteína C reactiva, estudio de médula ósea (incluyendo cartometría de flujo, citogenética e hibridación con florescencia), MRI y/o PET-CT.
Un valor de albumina bajo y una elevación de la beta2-microglobulina son considerados factores pronósticos. Hoy en día los estudios de hibridación con florescencia para detectar las mutaciones se utilizan para dividir a los pacientes en dos grupos: riesgo alto y riesgo estándar. Dependiendo del grupo de riesgo se indicará el tratamiento.

Tratamiento
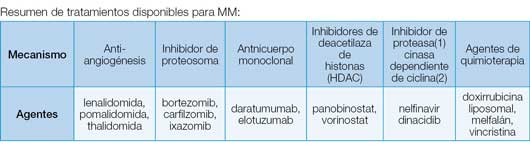
La terapia inicial de los pacientes con MM sintomático depende de la estratificación del riesgo, la elegibilidad para el trasplante autólogo de células hematopoyéticas y de los recursos disponibles. No existe un consenso general en cuanto al régimen de inducción preferido.
Los pacientes de alto riesgo reciben terapia de esteroides, un inmunomodulador (lenalidomida) y un inhibidor de proteasoma (bortezomib o carfilzomib). Esta triple terapia produce respuestas de hasta un 80%. Luego de 3 a 4 ciclos de tratamiento el paciente se considera para colección de células hematopoyéticas seguida de trasplante autólogo. Los pacientes que lo toleran reciben tratamiento de inmunomodulador o proteasoma de mantenimiento por tiempo indefinido.
Los pacientes de riesgo estándar se tratan de manera similar. Sin embargo, en este grupo se podrían coleccionar las células hematopoyéticas y guardarlas para hacer el trasplante autólogo cuando el paciente recaiga. La terapia de mantenimiento se puede comenzar luego de 6 a 8 ciclos del tratamiento inicial y es considerada en casi todos los pacientes.
Los pacientes en recaída deben tener una evaluación completa que incluya todas las pruebas de laboratorio y los estudios pertinentes. Algunos solo tienen una recaída química (proteína más alta) con elevación del pico monoclonal únicamente. Estos pacientes podrían recibir un régimen de salvamento menos agresivo. Los pacientes de recaída clínica necesitan tratamiento lo antes posible. Usualmente se prefiere las combinaciones que incluyan los anticuerpos monoclonales: daratumumab (en combinación con lenalidomida o bortezomib/carfilzomib y esteroides) o elotuzumab (pomalidomide y esteroides). El daratumumab es un anticuerpo monoclonal contra el antígeno CD38 presente en los linfocitos y las células de MM. Es muy activo y se podría usar como agente sencillo. El elotuzumab es un inhibidor de SLAMF7 que es una glicoproteína presente en las células de MM. Si se usa sólo, este fármaco no tiene actividad como agente, pero combinado con pomalidomida posee muy buena actividad para pacientes que han tenido su primera recaída.
Trasplante autólogo y alogénico en MM
El trasplante autólogo de células hematopoyéticas (TA) sigue siendo un componente clave de la terapia de MM en pacientes elegibles y puede incorporarse como parte de la terapia inicial o en el momento de la primera recaída, según la estratificación del riesgo. Para pacientes elegibles con MM, recomendamos incluirlo como parte del tratamiento inicial. La mayoría de los pacientes de menos de 78 años son candidatos para el TA. Varios investigadores han presentado estudios que evidencian el aumento de sobrevida en los pacientes que reciben este tratamiento.
En algunos casos se puede considerar el TA doble o trasplante tándem. Se define esto cuando se efectúa un TA seguido de un segundo trasplante dentro de los primeros 6 a 12 meses después del primero. Esta modalidad terapéutica se considera para algunos casos de pronóstico muy pobre. Un estudio evidenció la superioridad de sobrevida en los pacientes trasplantados.4 El TA es considerado como parte del tratamiento establecido para los pacientes de MM en buena condición física que no tengan enfermedad cardiovascular severa o problemas hepáticos serios.
Un pequeño grupo de pacientes con MM podría ser elegible para un trasplante alogénico (TAL), pero su rol en la terapia en MM sigue siendo experimental. A los pacientes jóvenes con MM refractario les brindamos la oportunidad de discutir los riesgos y beneficios de TAL con un especialista en trasplantes. La decisión de realizar trasplante en esta población se debe en gran medida a la refractariedad de MM y al riesgo que presente cada caso en particular. Dichos trasplantes deberían realizarse idealmente en el contexto de un ensayo clínico.
Terapia de “Car T Cells”
La terapia CAR T cells (Chimeric Antigen Receptor T-Cells) es un tipo de inmunoterapia antitumoral en la que se manipulan genéticamente los linfocitos T. Se ha utilizado esta terapia en MM con resultados prometedores. Simplificándolo, el procedimiento se realiza manipulando los linfocitos T (autólogos) del paciente de forma tal que reaccionen en contra de un antígeno particular de la malignidad, se expandan y luego se infundan al paciente (es aún un procedimiento experimental).
Trasplante autólogo en MM en Puerto Rico
El primer caso de trasplante autólogo para MM en Puerto Rico se efectuó en 1998. Luego, en nuestra institución se han realizado cerca de 100 trasplantes autólogos en pacientes de MM con muy buena sobrevida, además de haber reportado 7 casos que recibieron terapia de mantenimiento con talidomida luego del trasplante (en año 2000).1 Recientemente, otras 2 instituciones privadas en Puerto Rico también están efectuando trasplantes en MM.
Avances con nuevas sustancias
Además, un nuevo estudio fase 2 en pacientes de MM en recidiva y refractarios muestra resultados promisorios en algunos casos –en especial en pacientes con translocación (11;14)– que vienen siendo tratados con combinación oral de venetoclax (ya aprobado para ACL y LCC), carfilzomib y dexametasona.8 Los resultados a mediano y largo plazo determinarán su utilidad, así como la de otras combinaciones que vienen siendo estudiadas (como selenexor y dexametasona).9
Conclusión
En resumen, el MM es una enfermedad en la que se ha experimentado un crecimiento insospechado del conocimiento de sus fundamentos fisiopatológicos y de la gama de modalidades terapéuticas. La posibilidad de que estos pacientes puedan mejorar su calidad de vida está cada vez más cerca.
Referencias
- Castro J et al. Preliminary Experience in patients with MM HD Chemotherapy and AT on Thalidomide as Maintenance. Blood 2000: Vol. 96 (11); Suppl. 2, 5342.
- Review of 1027 patients with MM, Mayo Clinics Proc. 2003: 78;21.
- Attal M et al; 1996; 335(2):91.
- Palumbo A et al. NEJM 2014;371(10):89.5.
- Gay F et al. Lancet Onc 2015 Dec; 16(16):1617-29.
- Dimopoulos MA. NEJM 2018; 379:1811-22.
- Stadtmauer EA et al; CAR-T Cells in MM, Blood 2016.
- Costa LJ et al. ASH 2018: abstract 303.
- Chiari et al. ASH 2018: abstract 599.















