








Amiloidosis cardiaca:
Evaluación, diagnóstico y tratamiento

Sonia I. Vicenty-Rivera, MD
Cardióloga
Directora Clínica de Fallo Cardiaco
VA Caribbean Healthcare System
La amiloidosis es un trastorno infiltrativo causado por deposición de proteínas de bajo peso molecular en diferentes tejidos del cuerpo (amiloide).1 En el corazón causa daño estructural secundario a la presencia de los depósitos de amiloide, llevando a una cardiomiopatía restrictiva.2 Entre las diferentes subcategorías de amiloidosis, la mayoría de los casos de cardiomiopatías restrictivas (más del 95%) son por amiloidosis de transtiretina (ATTR) o amiloidosis de cadena ligera (AL).3
Aspectos clínicos
Los casos de amiloidosis cardiaca ATTR generalmente se presentan en pacientes de 60 años o más. Esta se subdivide, a su vez, en amiloidosis de tipo salvaje (wild-type ATTR o wt-ATTR) y hereditaria (h-ATTR). Ambas ocurren como resultado del depósito de transtiretina (TTR), la cual es una proteína sintetizada por el hígado y que transporta la hormona tiroidea y el retinol. Se describen varias mutaciones de transtiretina, asociadas con diferentes edades de presentación (varían entre los 30 y los 70 años) y con los riesgos de cardiomiopatía. Las manifestaciones clínicas son variadas, e incluyen estenosis espinal lumbar, síntomas gastrointestinales, nefropatía y arritmias, así como neuropatías autónomas y periféricas. La mutación más común de miocardiopatía a nivel mundial es la V122I, la cual tiene una incidencia entre afroamericanos y en pacientes de ascendencia caribeña entre el 3 y el 4%.4
Por otra parte, los casos de pacientes con amiloidosis cardíaca AL ocurren en personas de 40 años o más. Esta es causada por un trastorno multisistémico producido por células monoclonales que generan fragmentos de inmunoglobulinas anormales que se desdoblan y depositan en los tejidos como fibras amiloides. En esta condición existe compromiso cardiaco en un 50% a un 70% de los pacientes afectados, observándose una proporción igual en hombres y en mujeres. Su pronóstico es bastante pobre cuando no hay tratamiento.5
Aspectos diagnósticos
La evaluación diagnóstica de la amiloidosis cardiaca comienza con el examen clínico inicial para evaluar los síntomas y los signos cardiacos y extracardiacos, e incluye las pruebas de laboratorio iniciales, un electrocardiograma de 12 derivaciones y un ecocardiograma bidimensional.
En la ecocardiografía, la característica más distintiva es una discordancia entre el aumento del grosor en las paredes del ventrículo izquierdo y los cambios electrocardiográficos. En la electrocardiografía se describe usualmente una disminución del voltaje de la onda QRS. Sin embargo, esta característica tiene baja sensibilidad y la prevalencia de bajo voltaje varía notablemente con la etiología de la cardiomiopatía. Se ve con mayor frecuencia en los pacientes con amiloidosis AL (60%) que en los que están afectados por amiloidosis ATTR (20%).
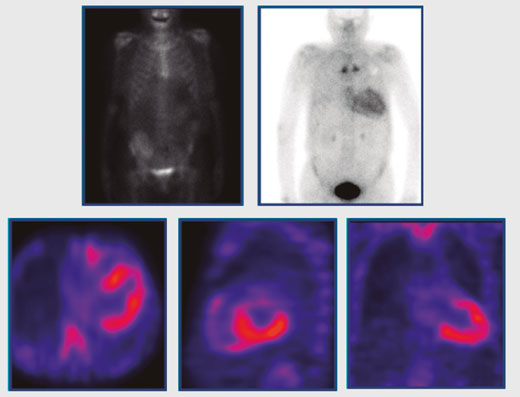
El enfoque diagnóstico se basa en la realización de una biopsia de tejido en el órgano afectado, en la que se demuestra evidencia de tinción roja de Congo (Apple Green Bridfringent). Para poder distinguir entre una amiloidosis cardíaca cardiaca tipo AL de una de tipo ATTR, se debe considerar el uso de un algoritmo que toma en cuenta los resultados de un estudio de medicina nuclear empleando pirofosfato marcado con 99m-Tecnecio (Tc99-PYP scan), y de las pruebas como ensayos de cadenas libres de suero, en conjunto con la inmunofijación de suero y orina para la detección de picos “M” de proteínas monoclonales. Este protocolo demostró una sensibilidad de hasta un 99% con una especificidad del 100% para diferenciar una amiloidosis cardiaca de tipo AL de una amiloidosis de tipo ATTR en pacientes sin evidencia de proteínas monoclonales y con un estudio Tc99-pirofosfato positivo.4
Opciones terapéuticas
La terapia para estos problemas tiene dos vertientes que consisten en el tratamiento y la estabilización de la insuficiencia cardiaca, al igual que en el manejo y la estabilización de la amiloidosis cardiaca. El manejo de la insuficiencia cardiaca en este grupo de pacientes es diferente del manejo que, por lo general, se recomienda para pacientes con fallo cardiaco debido a otras causas. Usualmente, estos pacientes tienden a ser intolerantes a agentes como los betabloqueadores, así como a los inhibidores de la convertasa de angiotensina. Del mismo modo, los bloqueadores de los canales de calcio que pueden ser útiles en el tratamiento del fallo diastólico son contraindicados en la cardiomiopatía amiloide. Se recomienda la anticoagulación en pacientes con miocardiopatía amiloide con fibrilación auricular, con algún trombo intracardiaco o con un evento embólico.
La principal opción de tratamiento en pacientes con amiloidosis AL es la quimioterapia con regímenes que incluyen al melfalano en dosis alta y con trasplante autólogo de células madre hematopoyéticas. Los regímenes basados en bortezomib se están convirtiendo en la terapia de primera línea, incluso en pacientes con enfermedad cardiaca avanzada (clase funcional III o IV).
Por otro lado, en la cardiomiopatía ATTR con NYHA (New York Heart Association Functional Classification) clase funcional I a III, se recomienda el tratamiento con tafamidis. Este es un estabilizador de la transtiretina que ha demostrado en estudios clínicos reducir la mortalidad y la necesidad de admisiones hospitalarias de estos pacientes.6
Comentario
La amiloidosis cardiaca es una enfermedad poco frecuente que suele afectar a pacientes a partir de la quinta década de vida. Luego de ser categorizada adecuadamente con las pruebas de diagnóstico recomendadas, se dispone en la actualidad de algunas terapéuticas –utilizando quimioterapias y anticuerpos monoclonales– que permiten reducir la mortalidad y prevenir sus complicaciones.
Referencias
- Siddiqi OK et al. Cardiac Amyloidosis: An Update on Pathophysiology, Diagnosis, and Treatment. Trends on Cardiovascular Medicine. 2018; 28(1): 10-21.
- Fontana M et al. Myocardial Amyloidosis: The exemplar Interstitial disease: JACC Cardiovasc Imaging 2019; 12:2345.
- Donnelly JP el al.CardiacAmyloidosis. An update on diagnosis and treatment. Cleveland Clinic Journal of Medicine 2017; 84:12-25.
- Ruberg FL et al. Transthyretin Amyloid Cardiomyopathy. JACC State-of the-Art Review. JACC. 2019; 73: 2872-91.
- Falk RH et al. AL (Light-Chain) Cardiac Amyloidosis: A review of Diagnosis and Therapy. JACC. 2016; 68(12): 1323.
- Maurer MS et al. Tafamidis Treatment for Patients with Transthyretin Amyloids Cardiomyopathy. NEJM 2018; 379:1007-1016.














