








Hipoglucemia en la edad pediátrica:
Evaluación y manejo para el clínico
Los primeros dos días después del alumbramiento, un recién nacido experimenta cambios fisiológicos en el metabolismo de la glucosa como transición para adaptarse a la vida extrauterina1. Luego de las primeras 48 horas de vida, el metabolismo de la glucosa adquiere bastantes similitudes con el del adulto.
Un grupo de expertos publicó hace poco una revisión del tema, de gran utilidad para el médico clínico2. Lo más importante son la identificación temprana y el manejo de todo paciente en que se sospeche hipoglucemia. Nuestro cerebro funciona en gran medida y en forma casi exclusiva por el suministro de glucosa (también en concentraciones adecuadas el lactato y los cuerpos cetónicos pueden servir como fuentes alternas de energía). Cuando persiste la hipoglucemia, hay riesgos de convulsiones y de daño cerebral permanente. De hecho, se estima que del 25 al 50% de los pacientes con hiperinsulinismo congénito pueden sufrir discapacidad debido a la hipoglucemia. En un adulto se estima que el 50% de la glucosa circulante es utilizada por el cerebro. El cerebro del recién nacido es relativamente más grande en comparación con el resto del cuerpo. Por esto, el consumo de glucosa del cerebro del recién nacido es proporcionalmente 2 a 3 veces mayor que el del adulto (4-6 mg/dl/min); esto lo predispone a un mayor riesgo cuando hay hipoglucemia.
La hipoglucemia clínica se define como la concentración de glucosa en plasma (GP) lo suficientemente baja como para causar síntomas y signos que impiden las funciones cerebrales, los que están bien delineados en la población adulta.
Los síntomas se dividen en:
- Neurogénicos (autonómicos; resultan de los cambios fisiológicos ocasionados por descargas del sistema simpático en respuesta a la hipoglucemia);
- Adrenérgicos (palpitaciones, temblor, ansiedad); y
- Colinérgicos (sudoración profusa, sensación de hambre y parestesias).
Los signos y síntomas de neuroglucopenia incluyen confusión, coma y convulsiones. El notar un episodio de hipoglucemia depende de que podamos percibir los efectos neurogénicos como respuesta a la hipoglucemia. La utilización de glucosa por el cerebro se hace limitante a un nivel de cerca de 55-65 mg% de GP. Los síntomas neurogénicos se suelen notar a una concentración de GP menor a 55 mg%; en niños más grandes y adultos esto dispara la respuesta de buscar comida o de pedir ayuda para evitar la hipoglucemia. Por otro lado, las funciones cognitivas (neuroglucopenia) se ven alteradas a niveles por debajo de 50 mg/% de GP.
Diagnóstico
Lo ideal es hacer el diagnóstico de hipoglucemia cumpliendo con la triada de Whipple:
- Nivel bajo de GP;
- Signos y síntomas de hipoglucemia; y
- Corrección de estos dos últimos al consumir o administrar una fuente de glucosa.
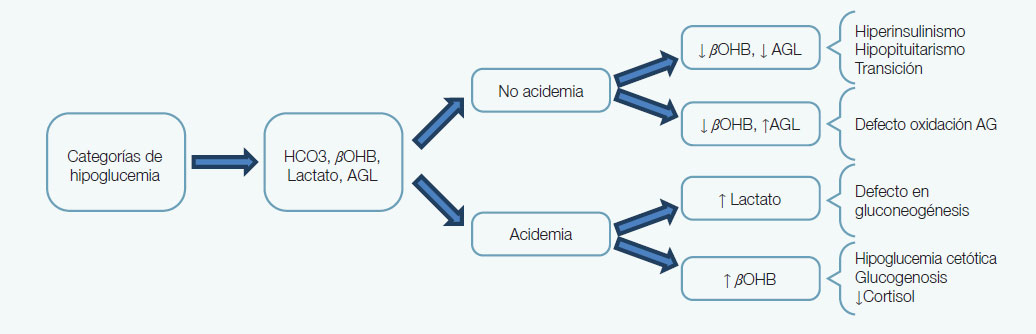
La hipoglucemia en infantes más grandes y niños es poco común, por lo que se debe insistir en identificar la triada de Whipple para evitar exponer al paciente a estudios innecesarios de poco o ningún beneficio, además de riesgosos y costosos. En neonatos, infantes y niños pequeños se debe confirmar el nivel bajo de GP para también evitar pruebas innecesarias. Además, la identificación de factores de riesgo y un alto índice de sospecha son claves para detectar la hipoglucemia en estos grupos. Para confirmar la hipoglucemia se pone énfasis en que el nivel de glucosa debe obtenerse en plasma tomando en cuenta posibles fuentes de error (e.g., GP disminuye a razón de 6mg/dl/hora si la muestra no es procesada en el momento). También, los métodos alternos para medir glucosa en sangre (por ejemplo los medidores de glucosa personales) pueden medir entre ±10-15 mg% menos que en plasma y pierden certeza en niveles bajos de glucosa en sangre. Idealmente, en cuanto se sospeche de hipoglicemia en un recién nacido, infante o adolescente se deben obtener muestras críticas que ayudarán a determinar la causa del evento (ver figura). En ausencia de las muestras críticas, se puede utilizar una prueba provocativa de ayuno prolongado. A grandes rasgos se desea separar estados de excesos de insulina de estado de poca o ninguna insulina. Los de exceso de insulina se caracterizan por ausencia de acidosis y de cuerpos cetónicos (beta hidroxibutirato; βOHB) con una baja concentración de ácidos grasos libres (AGL). Excepto en disturbios del metabolismo de oxidación de ácidos grasos en los que los AGL pudieran estar aumentados pero con poca o ninguna producción de cuerpos cetónicos (aun con niveles bajos de insulina no hay producción de cuerpos cetónicos). Por el contrario, en estados de baja o poca insulina hay acidemia y alta producción de lactato y βOHB (por ejemplo, en hipoglucemia cetótica).
Tratamiento
- El manejo inicial debe buscar restablecer el nivel de GP sobre 60 mg% con glucosa parenteral;
- Si se sospecha hiperinsulinismo (e.g., necesidad de infusión >8 mg de glucosa/kg/min) se puede administrar glucagón, que además del tratamiento puede servir de diagnóstico del hiperinsunilismo cuando se observa un incremento sobre 35 mg% en los niveles de GP tras la inyección por liberación de glucosa hepática previamente almacenada en forma de glucógeno (asumiendo que hay integridad en los mecanismos responsables de glucogenolisis);
- El historial y el examen físico son importantes para orientarnos sobre las posibles etiologías de la hipoglucemia y para identificar grupos en riesgo (pueden sufrir de hiperinsulinismo bebés grandes para edad gestacional o nacidos de madres diabéticas o que hayan experimentado estrés mayor en el parto);
- Un historial de embarazos previos con hiperinsulinismo apunta a posibles mutaciones que inactivan los canales de potasio/sulfonilurea de la célula beta pancreática, manteniéndola en constante secreción de insulina;
- La presencia de hepatomegalia con hipoglucemia, hipocetonemia, con acidosis láctica, aumento en triglicéridos y ácido úrico puede sugerir una enfermedad de almacenamiento de glucógeno (como GSD tipo I; enfermedad de Von Gierke)3;
- Se debe sospechar una deficiencia de la hormona de crecimiento en todo recién nacido con hipoglucemia y microfalo (menor de 2,5 cm);
- La posibilidad de administración exógena de insulina debe sospecharse en ocasiones. Si bien los niveles altos de insulina con niveles bajo de péptido C sugieren administración exógena de insulina, es importante conocer también que no todas las pruebas comerciales disponibles para medir la insulina pueden medir sus análogos;
- No se recomienda tratamiento empírico con esteroides a menos que se sospeche (y corrobore, en lo posible) la deficiencia de cortisol;
- El uso de diazoxide si puede ser ensayado y de provecho en pacientes con hiperinsulinismo;
- No debe haber ayuno prolongado para evitar recurrencia de episodios de hipoglucemia. Esta recurrencia se ha vinculado con poca o ninguna respuesta autonómica impidiendo la liberación de la glucosa hepática. A esto último se le conoce hoy en día como fallo autonómico asociado a hipoglucemia (FAAH). El FAAH puede ocurrir desde las primeras 10 a 13 semanas de vida;
- Antes de dar de alta a un paciente de alto riesgo de hipoglucemia, en particular si es infante, se debe asegurar que puede sostener niveles de GP sobre 70 mg% por al menos 6 a 8 horas;
- El tratamiento a largo plazo, independiente de la edad, se debe basar en la etiología de la hipoglucemia debiéndose buscar mantener un nivel de GP entre 70-100 mg% para evitar la activación neuroendocrina y el FAAH;
- Hay medicamentos para alguno de estos desórdenes, pero en ocasiones es necesaria la cirugía, como en el hiperinsulinismo intratable; y
- La terapia nutricional es la base del tratamiento de desórdenes de metabolismo de glucógeno.
Comentario
La hipoglucemia en la edad pediátrica es un desorden que debe de ser identificado cuanto antes para evitar compromisos neurológicos. Su evaluación y diagnóstico son importantes para instaurar la mejor opción terapéutica lo antes posible.
Referencias -# Stanley CA, Rozance PJ, Thornton PS, De Leon DD, Harris D, Haymond MW, et al. Re-evaluating “Transitional neonatal hypoglycemia”: mechanism and implications for management. J Pediatr 2015;166:1520-5. -# Thornton PS, Stanley CA, De Leon DD, et al. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J Pediatr. 2015 Aug;167(2):238-45. -# Weinstein DA, Toth KS, Wolfsdorf JE. Glycogen storage disease. In: Pediatric Endocrinology and Inborn Metabolic Errors. Eds. Sarafoglou K, Hoffamnn GF, Roth KS. 2009 McGraw-Hill pp.71-81.














